Human Fetal Organs¶
To prove the ability that SCRIP can be applied to diverse tissue types and infer the target genes of TRs, we applied SCRIP to a scATAC-seq dataset of human fetal organs that covers 14 different tissues (Domcke et al., Science, 2020). Data was downloaded from GEO with accession GSE149683.
We use the lung sample as an example to suggest the process of custom analysis.
SCRIP enrich -i example/fetal_organ/data/GSM4508936_lung.h5 -s hs -p example/fetal_organ/lung_SCRIP -t 32
library(Seurat)
library(ggplot2)
library(dplyr)
library(ComplexHeatmap)
library(RColorBrewer)
library(patchwork)
library(data.tree)
library(gridExtra)
library(rlist)
library(phangorn)
library(scales)
library(dendextend)
library(tidytree)
library(ggtree)
library(ape)
library(phylogram)
library(clusterProfiler)
library(org.Hs.eg.db)
library(igraph)
library(ggraph)
library(tidygraph)
enri<-read.table("example/fetal_organ/lung_SCRIP/enrichment/SCRIP_enrichment.txt",header=T) #25,0.5,30
enri_b<-t(enri)
head(enri_b)
seurat <- CreateSeuratObject(counts = enri_b, project = "lung")
seurat@assays$RNA@scale.data<-as.matrix(seurat@assays$RNA@counts)
seurat <- FindVariableFeatures(seurat, selection.method = "vst", nfeatures = 2000)
seurat <- RunPCA(seurat, features = VariableFeatures(object = seurat))
ElbowPlot(seurat)
seurat <- FindNeighbors(seurat, dims = 1:25)
seurat <- FindClusters(seurat, resolution = 0.7)
seurat <- RunUMAP(seurat, dims = 1:50)
clusters<-seurat@active.ident
clusters <- as.data.frame(clusters)
clusters$cell <- rownames(clusters)
a=c("Ciliated epithelial cells","Lymphatic endothelial cells","Lymphoid cells","Megakaryocytes","Myeloid cells",
"Neuroendocrine cells")
b=c("Stromal cells")
c=c("Bronchiolar and alveolar epithelial cells")
d=c("Vascular endothelial cells")
cluster_list <- list()
for (i in a){
clusters_cell <- clusters[which(clusters$clusters==i),"cell"]
cluster_list[[i]] <- sample(clusters_cell,ceiling(length(clusters_cell)/1))
}
for (i in b){
clusters_cell <- clusters[which(clusters$clusters==i),"cell"]
cluster_list[[i]] <- sample(clusters_cell,ceiling(length(clusters_cell)/20))
}
for (i in c){
clusters_cell <- clusters[which(clusters$clusters==i),"cell"]
cluster_list[[i]] <- sample(clusters_cell,ceiling(length(clusters_cell)/10))
}
for (i in d){
clusters_cell <- clusters[which(clusters$clusters==i),"cell"]
cluster_list[[i]] <- sample(clusters_cell,ceiling(length(clusters_cell)/2.5))
}
selected_cells <- unlist(cluster_list)
lung<-seurat
lung_use<-seurat[,selected_cells]
lung.markers <- FindAllMarkers(lung_use, only.pos = TRUE, min.pct = 0.1, logfc.threshold = 0.1)
lung.markers_use<-lung.markers %>%
group_by(cluster) %>%
slice_max(n = 30, order_by = avg_log2FC)
gene<-unique(lung.markers_use$gene)
mat <- GetAssayData(lung_use, slot = "counts")
mat_use<-as.matrix(mat[gene,])
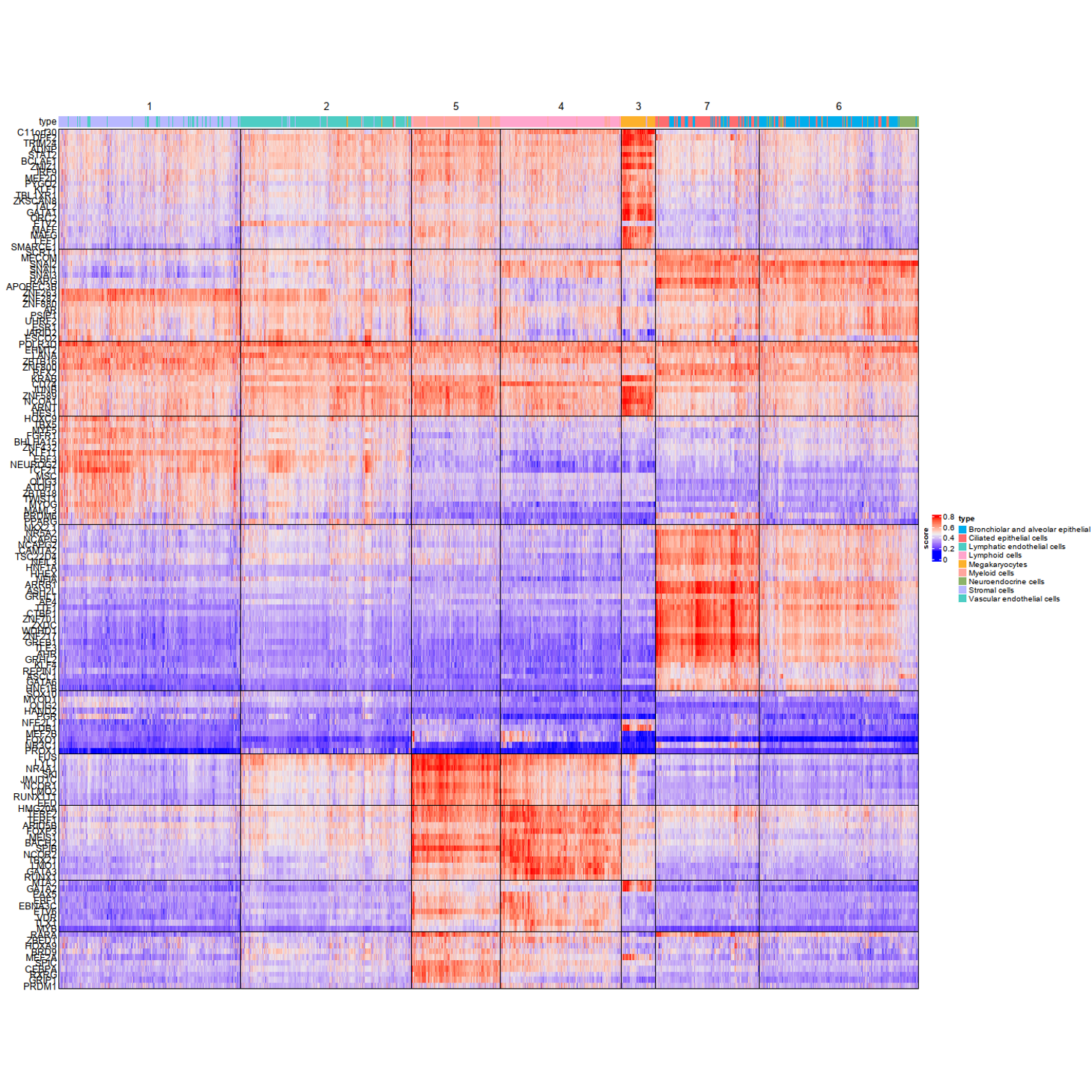
mycol=c("#FF6D6F","#00AEEC","#8cb369","#b8b8ff","#FEFBDD", "#FFA6CD","#cddafd","#4ecdc4","#FDB12C","#ffa69e")
names<-as.data.frame(lung_use@active.ident)
colnames(names)<-"cell_type"
type=names$cell_type
ha = HeatmapAnnotation(type = type, annotation_name_side = "left",
col=list(type=c("Bronchiolar and alveolar epithelial cells"=mycol[2],"Stromal cells"=mycol[4],"Vascular endothelial cells"=mycol[8],
"Lymphoid cells"=mycol[6],"Ciliated epithelial cells"=mycol[1],"Lymphatic endothelial cells"=mycol[8],
"Megakaryocytes"=mycol[9],"Myeloid cells"=mycol[10],"Neuroendocrine cells"=mycol[3])
))
for (i in 1:150){
min=min(mat_use[i,])
max=max(mat_use[i,])
for (c in 1:9417){
mat_use[i,c]=(mat_use[i,c]-min)/(max-min)
}
}
names$cell_type<-as.factor(names$cell_type)
annotation_col =names
options(repr.plot.width = 20, repr.plot.height = 20,repr.plot.res = 70)
set.seed(2021)
p<-Heatmap(mat_use,cluster_rows=TRUE,
cluster_columns=TRUE,
show_column_names=FALSE,
show_row_names=TRUE,
top_anno=ha,
column_km=7,
row_km = 10,
# row_km=6,
show_row_dend=FALSE,
show_column_dend=FALSE,
# right_annotation=row_anno,
heatmap_legend_param=list(
title="score",
title_position="leftcenter-rot"
),
row_gap = unit(0, "mm"),
column_gap = unit(0, "mm"),
border = TRUE,
width = unit(40, "cm"),
height = unit(40, "cm"),
row_names_side = "left"
)
p
SCRIP impute -i example/fetal_organ/data/GSM4508936_lung.h5 -s hs -p example/fetal_organ/lung_SCRIP -f h5ad --factor GATA3
SCRIP target -i example/fetal_organ/lung_SCRIP/imputation/imputed_GATA3.h5ad -s hs -o GATA3_target.h5ad
# GATA3 is expressed in Lymphoid
Lymphoid_cells<-subset(lung_use,idents=c('Lymphoid cells'),invert=FALSE)
# We did a extra step to convert h5ad to 10x mtx format for easily reading into R
GATA3<-Read10X("imputation/GATA3_10x", gene.column =1)
use_GATA3<-GATA3[,colnames(GATA3)%in%rownames(Lymphoid_cells@meta.data)] #choose lymphoid cell in GATA3 matrix
use_qc_GATA3<-use_GATA3[,colnames(use_GATA3)%in%rownames(as.data.frame(sort(colSums(use_GATA3),decreasing=TRUE)[1:500]))] #cell qc
Lymphoid_use_GATA3<-as.data.frame(sort(rowMeans(use_qc_GATA3),decreasing = TRUE)[1:1000]) #gene qc
eg_gata3_ly <- bitr(rownames(Lymphoid_use_GATA3), fromType="SYMBOL", toType=c("ENTREZID"), OrgDb="org.Hs.eg.db")
go_gata3_ly<-enrichGO(eg_gata3_ly$ENTREZID, OrgDb = org.Hs.eg.db, ont='BP',
pAdjustMethod = 'BH',
qvalueCutoff = 0.05,
keyType = 'ENTREZID')
